Genome assembly
Overview
Teaching: 45 min
Exercises: 30 minQuestions
How can I assemble a genome with the reads generated from the nanopore sequencers.
Objectives
Hybrid genome assembly using Nanopore and Illumina reads.
Scaffolding assembled contigs using RagTag.
Accessing genome assembly quality using BBMap and BUSCO.
Genome assembly
An overview of Genome Assembly
Once we have the “clean” Nanopore reads, we are ready to assemble those reads into contiguous sequences (contigs). There are many tools which deploy different algorithms to assemble genomes with nanopore reads. Due to the nature of the Long-Read sequencing technologies, the pipeline for assembling error-prone Nanopore reads incorporate error correction to facilitate identifying overlapping reads and improve assembly.
Genome assembly using SMARTdenovo
We will assemble the draft assembly using de novo assembler for our Nanopore reads. This software does not include error correction steps. SMARTdenovo deploys overlap-layout-consensus (OLC) algorithm.
Overlap-Layout-Consensus (OLC) algorithm
An assembly methods finds overlaps among all the reads, and then build the overlap graph to sort into the contigs. Align those contigs to make a consensus sequence.

SLURM scripts
To get started with SMARTdenovo, lets create new directory in your work directory and copy a submission script template from /blue/general_workshop/share/bash_files directory.
$ cd /blue/general_workshop/<username>
$ mkdir SMARTdenovo
$ cd SMARTdenovo
$ cp /blue/general_workshop/share/bash_files/Smartdenovo.sh ./Smartdenovo.sh
Note: .sh is commonly used extension for shell scripts. Using an extension is not mandatory.
Adding information to SLURM script
We have to modify some information in the template to provide more information to SLURM about the job.
We can use a small text editor program called nano to edit a file.
This will open a basic text editor.
$ nano Smartdenovo.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: Smartdenovo.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=Smartdenovo # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb # Job memory request
#SBATCH --time 12:00:00 # Time limit hrs:min:sec
#SBATCH --output=SuwSamrtdenovo_%j.out # Standard output and error log
pwd; hostname; date
module load smartdenovo/20180219
# Run genome assembly
smartdenovo.pl -p -t 1 Suw -c 1 /blue/general_workshop/share/Suwannee/Suw2_filtered_3000bp_60X.fastq > Suw.mak
make -f Suw.mak
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Editing in nano
nano is a command line editor. You can only move your cursor with arrow keys: ↑, ↓, ← and →. Clicking with mouse does not change the position of the cursor. Be careful, you may be editing in wrong place.
If you accidentally edited in wrong place, exit nano, delete the script
slurm.shand copy again from share directory.
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Checking usage of smartdenovo.pl
$ module load smartdenovo/20180219
$ smartdenovo.pl
Usage: smartdenovo.pl [options] <reads.fa>
Options:
-p STR output prefix [wtasm]
-e STR engine of overlaper, compressed kmer overlapper(zmo), dot matrix overlapper(dmo), [dmo]
-t INT number of threads [8]
-k INT k-mer length for overlapping [16]
for large genome as human, please use 17
-J INT min read length [5000]
-c INT generate consensus, [0]
Make: software build automation tool
The make program is an intelligent utility and works based on the changes you do in your source file (makefile), which is
Suw.mak. Makefile simplifies the process of building program.
Running a job in SLURM
Submitting a SMARTdenovo job
To submit the job to SLURM, sbatch command is used.
$ sbatch Smartdenovo.sh
Submitted batch job <jobid>
Checking status of a SLURM job
You can check the status of the job using the command squeue.
-u argument accepts <username> and displays all jobs by the user.
-A argument accepts account name and displays all jobs
using the resources allocated to that account.
$ squeue -u <username>
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
43807246 hpg-milan Smartden <username> R 0:14 1 c0709a-s3
$ squeue -A general_workshop
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
<jobid> hpg2-comp Smartden <user1> R 0:07 1 c29a-s2
<jobid> hpg2-comp Smartden <user2> R 1:32 1 c15a-s1
<jobid> hpg2-comp Smartden <user3> R 2:01 1 c09a-s4
Understanding Job Status
Under status
ST,Rstands for Running andPDstands for pending. If the job is pending, a reason may be provided in last column. Eg:
- None: Just taking a while before running.
- Priority: Higher priority jobs exist in this partition.
- QOSMaxCpuPerUserLimit: The user is already using max number of CPU that they are allowed to use.
Checking the log file
The SLURM submission script contains a line #SBATCH --output Samrtdenovo_%j.out. Thus, the output for this job with be in the file Samrtdenovo_<jobid>.log.
The job might take about a day, so we will not have the output by the end of today. Let’s copy the log file from /blue/general_workshop/share/Suwanee2/Smartdenovo directory.
$ cp /blue/general_workshop/share/Suwannee/Smartdenovo/SuwSmartdenovo_58583802.out ./SuwSmartdenovo_58583802.out
$ ls
Smartdenovo.sh Suw.fa.gz SuwSamrtdenovo_<jobid>.out
Suw.dmo.ovl Suw.mak SuwSmartdenovo_58583802.out
$ head SuwSmartdenovo_58583802.out
/blue/jeremybrawner/smithk/F_Cir/Suwannee2/Smartdenovo # wroking directory
c5a-s23.ufhpc # hostname (a label that is assigned to a device connected to a computer network)
Sat Sep 12 12:02:30 EDT 2020 (Date)
/apps/smartdenovo/20180219/bin/wtpre -J 5000 /blue/jeremybrawner/smithk/F_Cir/Suwannee2/Suw2_filtered_3000bp_60X.fastq | gzip -c -1 > Suw.fa.gz
/apps/smartdenovo/20180219/bin/wtzmo -t 8 -i Suw.fa.gz -fo Suw.dmo.ovl -k 16 -z 10 -Z 16 -U -1 -m 0.1 -A 1000
[Sat Sep 12 12:04:43 2020] loading long reads
[Sat Sep 12 12:05:39 2020] Done, 93129 reads (length >= 0)
[Sat Sep 12 12:05:39 2020] sorted sequences by length dsc
[Sat Sep 12 12:05:39 2020] calculating overlaps, 8 threads
[Sat Sep 12 12:05:39 2020] indexing 1/1
$ tail SuwSmartdenovo_58583802.out
103 tips, 7 bubbles, 0 chimera, 5 non-bog, 0 recoveries
[Sun Sep 13 07:02:30 2020] repair 115 bog elements
3 tips, 0 bubbles, 0 chimera, 1 non-bog, 0 recoveries
[Sun Sep 13 07:02:30 2020] repair 4 bog elements
0 tips, 0 bubbles, 0 chimera, 0 non-bog, 0 recoveries
[Sun Sep 13 07:02:30 2020] generated 2067 unitigs
[Sun Sep 13 07:02:30 2020] recover 16 edges inter unitigs
[Sun Sep 13 07:03:00 2020] output 21 independent unitigs
[Sun Sep 13 07:03:00 2020] Done
/apps/smartdenovo/20180219/bin/wtcns -t 8 Suw.dmo.lay > Suw.dmo.cns 2> Suw.dmo.cns.log
The end of log file provides an important information that Nanopore reads were assembled into 21 unitgs.
Unitig vs Contig
Contig is a set of overlapped DNA fragments. While unitig contains multiple contigs.
Polishing contigs using Racon
The SMARTdenovo does not include error correction/polishing steps. Racon aims to generate consensus sequences which is similar or better quality compared to the output generated from other assembly algorithms deploying error correction or consensus steps. Racon support data generated from different Long-read technologies.
Mapping of Nanopore reads to the assembly
To get started with Racon, we need to map the Nanopore reads to our genome assembly.
Burrows-Wheeler Alignment (BWA) tool aligns DNA fragments to genome assembly/reference genome. Three algorithms are available: BWA-backtrack (short-read alignment only), BWA-SW and BWA-MEM. To process the long reads, BWA-MEM is faster and more accurate than BWA-SW.
Let’s create new directory in your work directory and copy a submission script template from /blue/general_workshop/share/bash_files directory.
$ cd /blue/general_workshop/<username>
$ mkdir Polishing
$ cd Polishing
$ cp /blue/general_workshop/share/bash_files/bwa.sh ./bwa.sh
Once you copy the script to your working directory, we need to edit the script.
$ nano bwa.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: bwa.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=BWA # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb # Job memory request
#SBATCH --time 12:00:00 # Time limit hrs:min:sec
#SBATCH --output=Suw_index_%j.out # Standard output and error log
pwd; hostname; date
module load bwa/0.7.17
# First generate index file of assembly
bwa index /blue/general_workshop/share/Suwannee/Smartdenovo/Suw.utg.fa
# Align filtered Nanopore reads to the assembly
bwa mem -t 1 -x ont2d /blue/general_workshop/share/Suwannee/Smartdenovo/Suw.utg.fa \
/blue/general_workshop/share/Suwannee/Suw2_filtered_3000bp_60X.fastq > Suw.sam
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Checking usage of bwa mem
$ module load bwa
$ bwa mem
Usage: bwa mem [options] <idxbase> <in1.fq> [in2.fq]
Algorithm options:
-t INT number of threads [1]
Scoring options:
-x STR read type. Setting -x changes multiple parameters unless overridden [null]
pacbio: -k17 -W40 -r10 -A1 -B1 -O1 -E1 -L0 (PacBio reads to ref)
ont2d: -k14 -W20 -r10 -A1 -B1 -O1 -E1 -L0 (Oxford Nanopore 2D-reads to ref)
intractg: -B9 -O16 -L5 (intra-species contigs to ref)
Note: Due to the page size limitation, only partial options are shown.
A faster option for mapping
minimap2 has relaced BWA-MEM for PacBio or Nanopore reads alignment. minimap2 retains major features of BWA-MEM, but about 50 times faster, more accurate.
Submitting a BWA job
Before we submit a new job, we need to cancel your previous job.
$ squeue -u <username>
$ scancel <jobid>
$ squeue -u <username>
To submit the job to SLURM, sbatch command is used.
$ sbatch bwa.sh
Submitted batch job <jobid>
Constructing the index from the assembly only takes about a minute but alignment will take about an hours. Therefore, we will use pre-computed result to proceed to the next step.
Run Racon
Let’s copy a submission script template from /blue/general_workshop/share/bash_files directory.
Check your current working directory.
$ pwd
If for some reasons you are not at /blue/general_workshop/username/Polishing, navigate yourself to the right directory.
$ cd /blue/general_workshop/<username>/Polishing
$ cp /blue/general_workshop/share/bash_files/Racon.sh ./Racon.sh
Once you copy the script to your working directory, we need to edit the script.
$ nano Racon.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: Racon.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=Racon # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb # Job memory request
#SBATCH --time 12:00:00 # Time limit hrs:min:sec
#SBATCH --output=Racon_Suw_%j.out # Standard output and error log
pwd; hostname; date
module load racon
racon -t 1 /blue/general_workshop/share/Suwannee/Suw2_filtered_3000bp_60X.fastq \
/blue/general_workshop/share/Suwannee/Polishing/Suw.sam \
/blue/general_workshop/share/Suwannee/Smartdenovo/Suw.utg.fa\
> ./SuwRacon.fasta
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Checking usage of racon
$ module load racon
$ racon -h
usage: racon [options ...] <sequences> <overlaps> <target sequences>
#default output is stdout
<sequences>
input file in FASTA/FASTQ format (can be compressed with gzip)
containing sequences used for correction
<overlaps>
input file in MHAP/PAF/SAM format (can be compressed with gzip)
containing overlaps between sequences and target sequences
<target sequences>
input file in FASTA/FASTQ format (can be compressed with gzip)
containing sequences which will be corrected
options:
-t, --threads <int>
default: 1
number of threads
-h, --help
prints the usage
Submitting a Racon job
Before we submit a new job, we need to cancel your previous job.
$ squeue -u <username>
$ scancel <jobid>
$ squeue -u <username>
To submit the job to SLURM, sbatch command is used
$ sbatch Racon.sh
Submitted batch job <jobid>
Checking the log file
Correction will take about 30 minutes. Let’s copy the output file from /blue/general_workshop/share/Suwanee/Polishing directory.
$ cp /blue/general_workshop/share/Suwannee/Polishing/Racon_Suw_58629660.out ./Racon_Suw_58629660.out
$ ls
bwa.sh Racon_Suw_<jobid>.out Suw_index_<jobid>.out SuwRacon.fasta
Racon.sh Racon_Suw_58629660.out Suw_index_58628076.out Suw.sam
$ cat Racon_Suw_58629660.out
/blue/jeremybrawner/smithk/F_Cir/Suwannee2/mapping
c2a-s22.ufhpc
Mon Sep 14 21:03:13 EDT 2020
[racon::Polisher::initialize] loaded target sequences 0.962586 s
[racon::Polisher::initialize] loaded sequences 57.437520 s
[racon::Polisher::initialize] loaded overlaps 40.405221 s
[racon::Polisher::initialize] aligning overlaps [====================] 19.403566 s
[racon::Polisher::initialize] transformed data into windows 15.280284 s
[racon::Polisher::polish] generating consensus [====================] 1307.944380 s
[racon::Polisher::] total = 1442.492319 s
Racon will take about 30 minutes to finish correcting our assembly. We might not have enough time to finish the racon program. That is alright. We have also provided pre-computed output (corrected draft assembly) at /blue/general_workshop/share/Suwanee2/Polishing.
Assembly polishing using Pilon
Pilon is a software aims to automatically improve draft assembly. Pilon requires two inputs: a FASTA file of assembly and BAM file of Illumina reads aligned to the assembly. Pilon identifies inconsistencies between the assembly and reads. Pilon aims to improve the input assembly including: 1) Single base differences, 2) Small indels, 3) Larger indel or block substitution events, 4) Gap filling, and 5) Identification of local mis-assemblies. Outputs from Pilon tool are a FASTA file of improved assembly and optional VCF file to visualize the discrepancy between FASTA file of assembly and Illumina reads.
Polishing Racon-corrected assembly using Pilon
Three major steps are involved:
- Align Illumina reads to Racon-corrected assembly using BWA
- Convert SAM file to BAM file and sort the bam file based on reads position in the Racon assembly
- Correcting assembly using Pilon using sorted BAM file consisting of Illumina paired-end alignments, aligned to the Racon-corrected assembly.
Run Pilon
Let’s create new directory in your work directory and copy a submission script template from /blue/general_workshop/share/bash_files directory.
$ cd /blue/general_workshop/<username>/Polishing
$ mkdir Pilon
$ cd Pilon
$ cp /blue/general_workshop/share/bash_files/pilonRound1.sh ./pilonRound1.sh
Once you copy the script to your working directory, we need to edit the script.
$ nano pilonRound1.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: pilonRound1.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=Pilon # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb # Job memory request
#SBATCH --time 12:00:00 # Time limit hrs:min:sec
#SBATCH --output=SuwGenome_pilon_%j.out # Standard output and error log
pwd; hostname; date
module load bwa/0.7.17 samtools/1.15 pilon/1.24
# First create index of Racon-corrected assembly.
bwa index /blue/general_workshop/share/Suwannee/Polishing/SuwRacon.fasta
# Two steps here:
# 1. mapping illumina reads to Racon-corrected assembly
# 2. sorted output SAM file into BAM file sorted by the mapping evidence
bwa mem -t 1 /blue/general_workshop/share/Suwannee/Polishing/SuwRacon.fasta \
/blue/general_workshop/share/Suwannee/TrimFastX_R1.fq \
/blue/general_workshop/share/Suwannee/TrimFastX_R2.fq | \
samtools view - -Sb | samtools sort - -o pilon1.sorted.bam
# Create index of mapping evidence
samtools index pilon1.sorted.bam
# Telling JAVA how much memory it should use (8 GB)
export _JAVA_OPTIONS="-Xmx8g"
# Run pilon
pilon --genome /blue/general_workshop/share/Suwannee/Polishing/SuwRacon.fasta \
--fix all --changes --frags pilon1.sorted.bam --threads 1 --output Suw_pilon1 \
--outdir /blue/general_workshop/<username>/Polishing/Pilon
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Also, replace <username> to your username. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Checking usage of samtools view
$ module load samtools
$ samtools view
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
Output options:
-b, --bam Output BAM
General options:
-S Ignored (input format is auto-detected)
Note: Due to the page size limitation, only partial options are shown.
samtools sort
Usage: samtools sort [options...] [in.bam]
Options:
-o FILE Write final output to FILE rather than standard output
Note: Due to the page size limitation, only partial options are shown.
module load pilon
pilon --help
Usage: pilon --genome genome.fasta [--frags frags.bam] [--jumps jumps.bam] [--unpaired unpaired.bam]
[...other options...]
--genome genome.fasta
The input genome we are trying to improve, which must be the reference used
for the bam alignments. At least one of --frags or --jumps must also be given.
--frags frags.bam
A bam file consisting of fragment paired-end alignments, aligned to the --genome
argument using bwa or bowtie2. This argument may be specifed more than once.
--changes
If specified, a file listing changes in the <output>.fasta will be generated.
--fix fixlist
A comma-separated list of categories of issues to try to fix:
"snps": try to fix individual base errors;
"indels": try to fix small indels;
"gaps": try to fill gaps;
"local": try to detect and fix local misassemblies;
"all": all of the above (default);
"bases": shorthand for "snps" and "indels" (for back compatibility);
"none": none of the above; new fasta file will not be written.
Submitting a Pilon job
Racon should be done by now. Check the job status first.
Before we submit a new job, we need to cancel your previous job.
$ squeue -u <username>
$ scancel <jobid>
$ squeue -u <username>
To avoid any potential error. We will submit the script using the Racon-corrected assembly computed by us previously.
To submit the job to SLURM, sbatch command is used
$ sbatch pilonRound1.sh
Submitted batch job <jobid>
Further polishing Pilon assembly using Pilon
We polished the Pilon assembly two times. We mapped Illumina reads to 1st Pilon-polished assembly to obtained 2nd Pilon-polished assembly. Pilon program takes about 30 minutes. We have prepared the 2nd Pilon-polished assembly for the next step.
Scaffolding: mapping the polished assembly to the reference genome
RagTag is a collection of software tools for scaffolding and improving genome assemblies. RagTag performs:
- Homology-based misassemble correction
- Homology-based assembly scaffolding and patching
- Scaffold merging
In our case, we will use the scaffolding function.
RagTag
- Correction: RagTag identifies and correct potential misassembles in the query assembly using reference genome.
- Scaffold: RagTag orders and orients sequences of draft query assembly into longer sequences using reference genome. Sequences will be jointed with gap without altering the sequences.
- Patch: RagTag fills the gaps in the query assembly using reference genome.
- Merge: RagTag merges different scaffoldings of the same query assembly, which can improve scaffolding using different scaffolding tools.
Run RagTag
First, create a RagTag folder under /blue/general_workshop/<username>/Polishing, then copy script from /blue/general_workshop/share/bash_files
$ cd /blue/general_workshop/<username>/Polishing
$ mkdir RagTag
$ cd RagTag
$ cp /blue/general_workshop/share/bash_files/ragtag.sh ./ragtag.sh
Adding information to SLURM script
We have to modify some information in the template to provide more information to SLURM about the job.
$ nano ragtag.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: ragtag.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=RagTag # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=1GB # Job memory request
#SBATCH --time 1:00:00 # Time limit hrs:min:sec
#SBATCH --output=Ragtag_%j.out # Standard output and error log
module load ragtag/2.0.1
ragtag.py scaffold /blue/general_workshop/share/GCA_000497325.3/GCA_000497325.3_OldRef.fna \
/blue/general_workshop/share/Suwannee/Polishing/pilon_round2/Suw_pilon2.fasta
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Running a job in SLURM
Submitting a RagTag job
Before we submit a new job, we need to cancel your previous job.
$ squeue -u <username>
$ scancel <jobid>
$ squeue -u <username>
To submit the job to SLURM, sbatch command is used.
$ sbatch ragtag.sh
Submitted batch job <jobid>
RagTag only takes a minutes, so let’s check the output in /blue/general_workshop/<username>/Polishing/Ragtag/ragtag_output.
$ cd /blue/general_workshop/<username>/Polishing/Ragtag/ragtag_output
$ ls
ragtag.scaffold.agp ragtag.scaffold.confidence.txt ragtag.scaffold.stats
ragtag.scaffold.asm.paf ragtag.scaffold.err
ragtag.scaffold.asm.paf.log ragtag.scaffold.fasta
Outputs of RagTag
- ragtag.scaffold.agp: The order and orientation of query assembly in AGP format.
- ragtag.scaffold.fasta: Scaffold in FASTA format, defined by the ragtag.scaffold.agp
- ragtag.scaffold.stats: Summary statistics for the scaffolding process.
Let’s look at the statistics report.
cat ragtag.scaffold.stats
placed_sequences placed_bp unplaced_sequences unplaced_bp gap_bp gap_sequences
19 46269058 2 917303 600 6
Quality assessment of assemblies
Computing metrics of assemblies using QUAST
To perform assembly evaluation, we will run QUAST. QUAST computes several common metrics including misassembles, contig N50, genome fraction (aligned based in the assemblies/reference genome). QUAST provides several outputs including report.txt, assessment summary in plain text file, and HTML file, a report including interactive plots. You can read the complete manual here.
Run QUAST
First, create a QUAST folder at /blue/general_workshop/<username>, then copy a submision script from /blue/general_workshop/share/bash_files
$ cd /blue/general_workshop/<username>
$ mkdir QUAST
$ cd QUAST
$ cp /blue/general_workshop/share/bash_files/quast.sh ./quast.sh
Adding information to SLURM script
We have to modify some information in the template to provide more information to SLURM about the job.
$ nano quast.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: quast.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=QUAST # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=1gb # Job memory request
#SBATCH --time 1:00:00 # Time limit hrs:min:sec
#SBATCH --output=QUAST_%j.out # Standard output and error log
pwd; hostname; date
module load quast
quast.py /blue/general_workshop/plyu/Polishing/Ragtag/ragtag_output/ragtag.scaffold.fasta \
/blue/general_workshop/share/GCA_000497325.3/GCA_000497325.3_OldRef.fna \
/blue/general_workshop/share/GCA_024047395.1/GCA_024047395.1_New_Ref.fna \
-o quastResult
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Fusarium circinatum has 21 assemblies available on NCBI Assembly. We will compared two most recent uploaded assemblies: GCA_000497325.3 (Uploaded in 2018; previous reference genome) and GCA_024047395.1 (Uploaded in July 2022; current reference genome).
GCA_000497325.3 assembly was assembled from reads generated by ABI Solid Sequencing and 454 (Next-generation seducing technologies); GCA_024047395.1 is a hybrid assembly using PacBio technology and Illumina HiSeq.
Running a job in SLURM
Submitting a QUAST job
To submit the job to SLURM, sbatch command is used.
$ sbatch quast.sh
Submitted batch job <jobid>
QUAST analysis will only take couple minutes, so we let’s look at the outputs.
$ ls
QUAST_<jobid>.out quastResult quast.sh
$ cd quastResult
$ ls
basic_stats report.html transposed_report.tex
icarus.html report.pdf transposed_report.tsv
icarus_viewers report.tex transposed_report.txt
QUAST_43889307.out report.tsv
quast.log report.tx
Let’s download report.html file. On your dashboard of UFRC OnDemand:

- Click Files, then /blue/general_workshop.
- Find your folder, click QUAST, then click quastResult.
- Select report.html, then click
Download.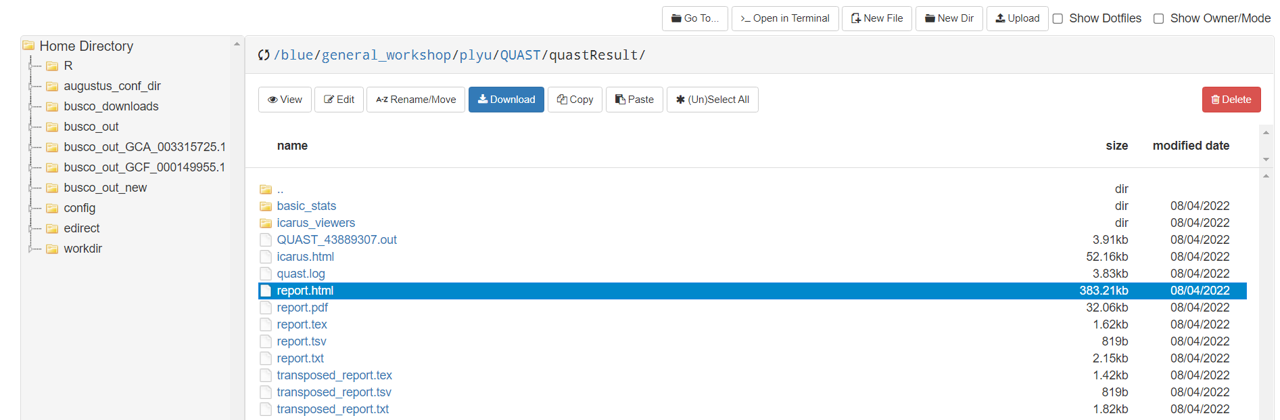
Open the HTML file locating in your local folder.
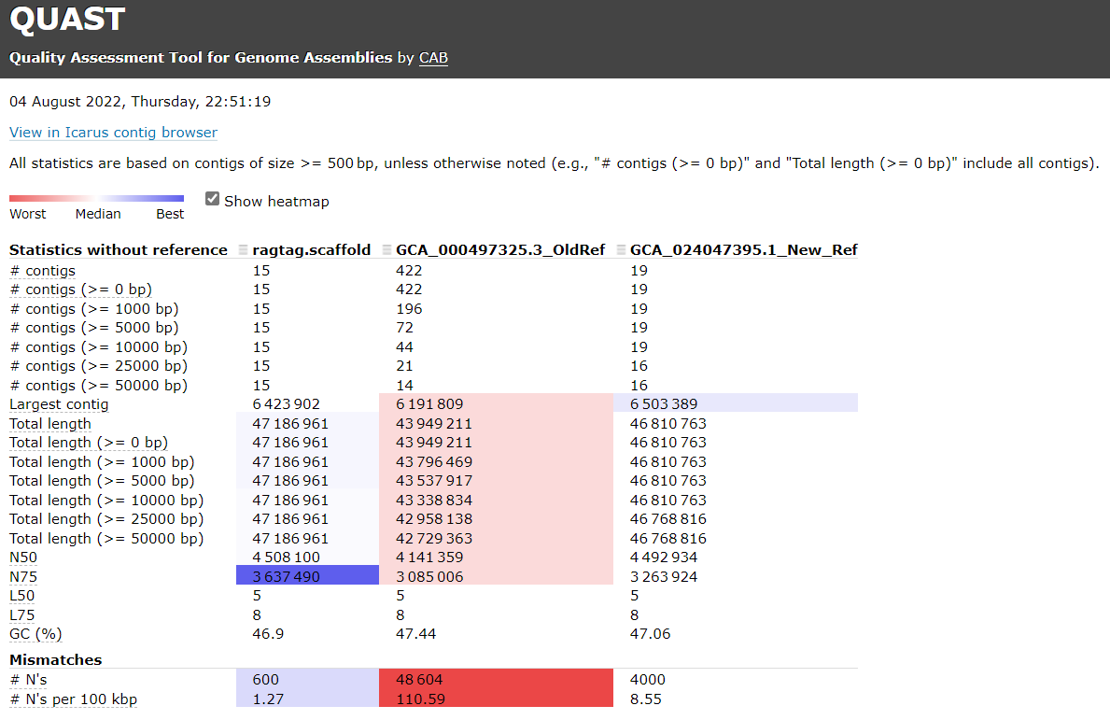
Evaluate assembly completeness using BUSCO
A measure for quantitative assessment of genome assembly and annotation completeness based on evolutionarily informed expectations of gene content was proposed. A oopen-source software, with sets of Benchmarking Universal Single-Copy Orthologs, named BUSCO, is available (Simao et al., 2015).
Run BUSCO analysis
First, create a BUSCO folder under your folder, then copy the configuration of BUSCO containing all the required dependencies from /blue/general_workshop/share/BUSCO/augustus.
$ cd /blue/general_workshop/<username>
$ mkdir BUSCO
$ cd BUSCO
$ cp -r /blue/general_workshop/share/BUSCO/augustus ./augustus
$ cp /blue/general_workshop/share/bash_files/busco.sh ./busco.sh
Adding information to SLURM script
We have to modify some information in the template to provide more information to SLURM about the job.
$ nano busco.sh
-----------------------------------------------------------------------------------------------
GNU nano 2.3.1 File: busco.sh
-----------------------------------------------------------------------------------------------
#!/bin/bash
#SBATCH --job-name=BUSCO # Job name
#SBATCH --account=general_workshop # Account to run the computational task
#SBATCH --qos=general_workshop # Account allocation
#SBATCH --mail-type=END,FAIL # Mail events (NONE, BEGIN, END, FAIL, ALL)
#SBATCH --mail-user=<email_address> # You need provide your email address
#SBATCH --ntasks=1 # Run on a single CPU
#SBATCH --cpus-per-task=1
#SBATCH --mem=2gb # Job memory request
#SBATCH --time 1:00:00 # Time limit hrs:min:sec
#SBATCH --output=BUSCO_%j.out # Standard output and error log
pwd; hostname; date
module load busco/5.3.0
busco -f -i /blue/general_workshop/share/Suwannee/Polishing/ragtag/ragtag_output/ragtag.scaffolds.fasta\
-o BUSCO_out_Suw --lineage_dataset hypocreales_odb10 -m genome
-----------------------------------------------------------------------------------------------
^G Get Help ^O WriteOut ^R Read File ^Y Prev Page ^K Cut Text ^C Cur Pos
^X Exit ^J Justify ^W Where Is ^V Next Page ^U UnCut Text ^T To Spell
-----------------------------------------------------------------------------------------------
Change the <email_address> to your email address where you can check email. Once you are done, press Ctrl+x to return to bash prompt. Press Y and Enter to save the changes made to the file.
Checking usage of busco
$ module load busco/5.3.0
$ busco -h
usage: busco -i [SEQUENCE_FILE] -l [LINEAGE] -o [OUTPUT_NAME] -m [MODE] [OTHER OPTIONS]
optional arguments:
-i FASTA FILE, --in FASTA FILE
Input sequence file in FASTA format. Can be an assembled genome or transcriptome (DNA), or protein sequences from an annotated gene set.
-o OUTPUT, --out OUTPUT
Give your analysis run a recognisable short name. Output folders and files will be labelled with this name. WARNING: do not provide a path
-l LINEAGE, --lineage_dataset LINEAGE
Specify the name of the BUSCO lineage to be used.
-m MODE, --mode MODE Specify which BUSCO analysis mode to run.
There are three valid modes:
- geno or genome, for genome assemblies (DNA)
Running a job in SLURM
Submitting a BUSCO job
To submit the job to SLURM, sbatch command is used.
$ sbatch busco.sh
Submitted batch job <jobid>
BUSCO analysis will take about an hour, so we prepared the pre-computed output.
We will copy the output of BUSCO analyses from our assembly, reference genome (GCA_024047395.1) and previous reference genome available on NCBI (GCA_000497325.3).
$ cp /blue/general_workshop/share/BUSCO/BUSCO_out_Suw/short_summary.specific.hypocreales_odb10.BUSCO_out_Suw.txt ./BUSCO_out_Suw.txt
$ cp /blue/general_workshop/share/BUSCO/BUSCO_out_Fc_ref/short_summary.specific.hypocreales_odb10.BUSCO_out_Fc_ref.txt ./BUSCO_out_Fc_ref.txt
$ cp /blue/general_workshop/share/BUSCO/BUSCO_out_Fc_oldref/short_summary.specific.hypocreales_odb10.BUSCO_out_Fc_oldref.txt ./BUSCO_out_Fc_oldref.txt
$ ls
augustus
busco.sh
BUSCO_<username>.out
BUSCO_out_Fc_oldref.txt
BUSCO_out_Fc_ref.txt
BUSCO_out_Suw.txt
$ cat BUSCO_out_Suw.txt
# BUSCO version is: 5.3.0
# The lineage dataset is: hypocreales_odb10 (Creation date: 2020-08-05, number of genomes: 50, number of BUSCOs: 4494)
# Summarized benchmarking in BUSCO notation for file /blue/general_workshop/share/Suwannee/Polishing/ragtag/ragtag_output/ragtag.scaffolds.fasta
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:98.0%[S:97.7%,D:0.3%],F:0.3%,M:1.7%,n:4494
4401 Complete BUSCOs (C)
4389 Complete and single-copy BUSCOs (S)
12 Complete and duplicated BUSCOs (D)
12 Fragmented BUSCOs (F)
81 Missing BUSCOs (M)
4494 Total BUSCO groups searched
Dependencies and versions:
hmmsearch: 3.1
metaeuk: 5.34c21f2
$ cat BUSCO_out_Fc_oldref.txt
# BUSCO version is: 5.3.0
# The lineage dataset is: hypocreales_odb10 (Creation date: 2020-08-05, number of genomes: 50, number of BUSCOs: 4494)
# Summarized benchmarking in BUSCO notation for file /blue/general_workshop/share/BUSCO/GCA_000497325.3/GCA_000497325.3_old_ref.fna
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:94.0%[S:93.9%,D:0.1%],F:3.1%,M:2.9%,n:4494
4225 Complete BUSCOs (C)
4219 Complete and single-copy BUSCOs (S)
6 Complete and duplicated BUSCOs (D)
139 Fragmented BUSCOs (F)
130 Missing BUSCOs (M)
4494 Total BUSCO groups searched
Dependencies and versions:
hmmsearch: 3.1
metaeuk: 5.34c21f2
$ cat BUSCO_out_Fc_ref.txt
# BUSCO version is: 5.3.0
# The lineage dataset is: hypocreales_odb10 (Creation date: 2020-08-05, number of genomes: 50, number of BUSCOs: 4494)
# Summarized benchmarking in BUSCO notation for file /blue/general_workshop/share/BUSCO/GCA_024047395.1/GCA_024047395.1_assembly_ref.fa
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:97.6%[S:97.3%,D:0.3%],F:0.6%,M:1.8%,n:4494
4384 Complete BUSCOs (C)
4372 Complete and single-copy BUSCOs (S)
12 Complete and duplicated BUSCOs (D)
28 Fragmented BUSCOs (F)
82 Missing BUSCOs (M)
4494 Total BUSCO groups searched
Dependencies and versions:
hmmsearch: 3.1
metaeuk: 5.34c21f2
References and additional reading
Key Points
Don’t forget to edit the email in the SLURM scripts and username.
There are several other ways to submit the job. Some software support multithread and you can write the array scripts request resources.