Basecalling and quality control
Overview
Teaching: 10 min
Exercises: 25 minQuestions
How do you perform basecalling and filter reads for analysis?
Objectives
Learn about basecalling with Guppy.
Learn how to use Porechop to remove adapter sequences.
Learn how Filtlong improves the quality of your data.
Use Nanoplot to produce summary reports of your dataset.
Basecalling and quality control
Basecalling
Basecalling with Guppy
The application Guppy converts the fast5 files we viewed earlier into fastQ files that we can use for bioinformatics applications. It is strongly recommended that you allocate a GPU when running this application. We know a researcher who used Guppy for basecalling while only using CPUs, which took 2-4 days to process their Nanopore results. However, when they used a GPU it only took 1-2 hours to process the same results.
Whether you use a CPU or GPU, 0’s and 1’s are interpreted by each processing core in a linear fashion. However, CPUs typically have 4-8 cores that are very powerful and can execute a relatively large number of tasks. GPUs, on the other hand, usually have at least 300 cores upwards to a thousand that are less powerful and can execute more simplistic and repetitive tasks. Since processing the fast5 electrical signals into nucleotides ATGC is a very simple and repetitive task, a GPU will outperform the CPU by a very large margin, although a CPU will work (much more slowly) if your work group does not have access to a GPU on the cluster.
You can include a GPU in your SLURM script using #SBATCH --partition=gpu and #SBATCH --gpus=<number> where number is how many GPUs you want to allocate. Since we are using a GPU we will also load the application CUDA, which facilitates the parallelization of processing data with a GPU to increase performance. We can see these options and more in the Guppy script below. First, we will run guppy on a subset of samples and review the script while it is running:
cd demo
sbatch guppy.sh
cat guppy.sh
#!/bin/bash
#SBATCH --job-name=guppy
#SBATCH --account=general_workshop
#SBATCH --qos=general_workshop
#SBATCH --time=24:00:00
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb
#SBATCH --mail-type=BEGIN,END,FAIL,TIME_LIMIT
#SBATCH --mail-user=USERNAME@ufl.edu
#SBATCH --output=guppy_%j.out
#SBATCH --error=guppy_%j.err
#SBATCH --partition=gpu
#SBATCH --gpus=1
date;hostname;pwd
module purge
module load cuda/11.0.207 guppy/4.4.1
#Before basecalling, create a folder names basecall_out
mkdir ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out
guppy_basecaller \
--recursive \
--input_path ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/fast5 \
--save_path ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out \
-c dna_r9.4.1_450bps_hac.cfg \
-x cuda:0
## Then we concatenate all the fastq files into one big file
cat ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out/*.fastq \
> ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out/bigfile.fastq
Let’s review some of the options:
--recursivetells guppy to search for more than one file in the folder and proceed through all of them.--input_pathis the location with the fast5 files--save_pathis the location the saved files should be written to-cis the configuration file indicating what flowcell and kit were used for sequencing. We will discuss this more below.-xspecifies the basecalling device, which is a GPU using cuda in this case,cuda:0.
Guppy will save each fast5 file as a corresponding fastq file during basecalling. You can lump all of these results into a single fastq file at the end of basecalling using the final `cat` command, which uses a wildcard to write all of the files with the `*.fastq` extension to a single fastq file. This can make processing the data much easier downstream because you will only need to reference one file instead of an entire folder of files.
You may use a different type of flowcell or sequencing kit for your own work in the future than what is listed above. You can load the guppy application and run a command to list all of the available flowcell and kit combinations to determine which configuration file name to use. This list is very long, so we will pipe the results to the | head command and list the first few options.
module load guppy/6.1.7
guppy_basecaller --print_workflows | head
Loading model version information, please wait .................................................................
Available flowcell + kit combinations are:
flowcell kit barcoding config_name model version
FLO-FLG001 SQK-RNA001 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-FLG001 SQK-RNA002 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-MIN106 SQK-RNA001 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-MIN106 SQK-RNA002 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-MINSP6 SQK-RNA001 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-MINSP6 SQK-RNA002 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
FLO-MIN107 SQK-RNA001 rna_r9.4.1_70bps_hac 2020-09-07_rna_r9.4.1_minion_256_8f8fc47b
Note that when we load modules we load specific versions of the applications
cuda/11.0.207andguppy/4.4.1. When you publish your results, you will need to indicate which version of an application you used. HPCs will routinely update applications without notice, so you could run a different version of the basecaller the same day, which could potentially have an effect on the results. You can look up which versions of an application are currently available on the HPC using the commandmodule spiderfollowed the the name of the application.
module spider guppy
----------------------------------------------------------------------------------------------------------------------------------------------------------
guppy:
----------------------------------------------------------------------------------------------------------------------------------------------------------
Description:
A basecaller for Oxford Nanopore Technologies
Versions:
guppy/3.2.4
guppy/3.4.1
guppy/3.4.4
guppy/3.5.1
guppy/3.6.0
guppy/3.6.1
guppy/4.0.11
guppy/4.0.15
guppy/4.2.2
guppy/4.4.1
guppy/4.5.4
guppy/6.1.7
----------------------------------------------------------------------------------------------------------------------------------------------------------
For detailed information about a specific "guppy" package (including how to load the modules) use the module's full name.
Note that names that have a trailing (E) are extensions provided by other modules.
For example:
$ module spider guppy/6.1.7
----------------------------------------------------------------------------------------------------------------------------------------------------------
guppy_basecaller --help | head -n 25
: Guppy Basecalling Software, (C) Oxford Nanopore Technologies plc. Version 6.1.7+21b93d1, minimap2 version 2.22-r1101
Use of this software is permitted solely under the terms of the end user license agreement (EULA).By running, copying or accessing this software, you are demonstrating your acceptance of the EULA.
The EULA may be found in /apps/guppy/6.1.7/bin
Usage:
With config file:
guppy_basecaller -i <input path> -s <save path> -c <config file> [options]
With flowcell and kit name:
guppy_basecaller -i <input path> -s <save path> --flowcell <flowcell name>
--kit <kit name>
List supported flowcells and kits:
guppy_basecaller --print_workflows
Command line parameters:
-c [ --config ]
Configuration file for application.
-d [ --data_path ]
Path to use for loading any data files the application requires.
-x [ --device ]
Specify GPU device: 'auto', or 'cuda:<device_id>'.
-s [ --save_path ]
Path to save output files.
--ping_url
Finally, before moving on we can take a quick look at one of the fastq files with the results that we can now read as the nucleotide sequence and coding for the quality of each basecall:
head /blue/general_workshop/share/Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out/fastq_runid_0b0467cebb629a6562626fabc8c6546dc10cfdd5_229_0.fastq -n 4
@0792e4aa-713a-478f-ab5c-7eecab8fc118 runid=0b0467cebb629a6562626fabc8c6546dc10cfdd5 sampleid=Suwanee_2ndRun read=15586 ch=391 start_time=2020-08-09T03:15:06Z
AGTATGCTTCGTTCAGTTACGTATTGCTCGGCTGATCTGAGCTTGTAAAAGAGAATCGATGTGGCATATGTGGATAGCATGGGAAACGGCTTTTTCTTGAGAGAACCACGTCACCTCGATTTCGCCAGCTTTCCCAAGCTTTACAATGCTTGTGGTGTCTCCAATTGCGCTGTATGGTCACAGGAACAAACCGCACTTACTATATTTCCCATGTTTGCCGTCTTCATTGGACTCGAGCTTCTGGGTTGGCCGAAAACCAAGGACATTGATACTCAGTCAAGTGTGGAAGCTGACCATAGCGGCAGCAAGGAAGGTCCGGAGTCTTAACGTATGCGGAGAGTCAGGTACTCAAACAGCTAAGGCTACGCCTGAGGATACAATTTTCCAGACGTTTGCTACTTAATACGTAACTTG
+
%''(%%:;<9>@G3/;BAADCC<<?;997/3;:984,)))278;<<>?3%-7%:%>7<??=:>:CBCGA<F9;?8=;=890.,>DA?*0;?BFC?<033,%+''<:<8<8??:;7;-,,.13/'&%2'8<@C998236<AB>10<7=8((.-4558:96:89;=AEG>:?@:8/24957-+'<7:A=>>:A@7=?>A?I?BDFH?CI@A=DE?EKGB89>B:=AABCG=:A;?8:959?@8B?==@HC=778;B=<=?FH959;EFEBAED;H;<=AA).2?C@@C<;;?@AD=?9<8;=?A::9>6?890(0/00/*)$(2&.1799=AA9>=>@?85;;3/:6A:5<7=??@AB?;A=9<>=7;989:,%(1153;<>=?=8/3123/,36+&)22777?-744)++)$$%$
Filtering the basecaller results
Removing adapters with Porechop
During library preparation, you attach adapters to the nucleotide strands in your sample to prepare them for sequencing. Since these are not part of the original sequences, we will remove them using Porechop.
sbatch porechop.sh
cat porechop.sh
#!/bin/bash
#SBATCH --account=general_workshop
#SBATCH --qos=general_workshop
#SBATCH --job-name=porechop
#SBATCH --output=porechop_%j.out
#SBATCH --error=porechop_%j.err
#SBATCH --mail-type=END,FAIL
#SBATCH --mail-user=jkonkol@ufl.edu
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb
#SBATCH --time=96:00:00
pwd; hostname; date
module load gcc/5.2.0 porechop/0.2.4
porechop -i ./Suwannee2/20200808_1457_MN33357_FAL75110_031a874e/basecall_out/bigfile.fastq \
-o ./Suwannee2/PorechopOutSuw2.fastq -t 1
Note that we have loaded the module gcc, which is a C++ compiler. A C++ compiler is required to load and use porechop, so be sure to include it in your script.
Let’s review the options we used:
-iis the input fastq file-ois the output fastq file for the filtered results-tis the number of threads or CPUs assigned for the task. You can include more for multithreading if you allocate additonal CPUs for the job.- Porechop includes many other options you might be interested in using. You can review these on their github support site: [Porechop]https://github.com/rrwick/Porechop#install-from-source{: target=”_blank”}.
Now that Porechop is finsihed we can filter our reads for quality.
Filtering results with Filtlong
We can filter our results for both length and quality using the application Filtlong. Let’s take a look at the script:
sbatch filtlong.sh
cat filtlong.sh
#!/bin/bash
#SBATCH --job-name=filtlong
#SBATCH --account=general_workshop
#SBATCH --qos=general_workshop
#SBATCH --mail-type=END,FAIL
#SBATCH --mail-user=jkonkol@ufl.edu
#SBATCH --time=24:00:00
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --mem=8gb
#SBATCH --output=filtlong_%j.out
#SBATCH --error=filtlong_%j.err
pwd; hostname; date
module load filtlong/0.2.0
filtlong --min_length 3000 --target_bases 6000 ./Suwannee2/PorechopOutSuw2.fastq \
> ./Suwannee2/Suw2_filtered_3000bp_60X.fastq
# --min_length minimum length threshold
# --target_bases keep only the best reads up to this many total bases
Filtlong can filter the quality of de novo reads (the method used in this pipeline) based upon their length and the quality of its basecalls. Here is a review of the options that we used:
--min_lengthwill discard any reads shorter than a specific length. Generally this value is set to 1000 or higher.--target_baseswill remove the worst reads in the dataset until there are fewer than this threshold (it has no effect if the input has fewer than this number of reads).- These options are followed by the location of the fastq sequences and the location where the output is written.
The --target_bases option assumes that there is very high coverage of the genome from the sequencing results, and the threshold 3600000000 above was used to give about 60X coverage. If sequencing coverage is lower, you can use the option keep_percent 90, which will remove the worst 10% of the reads. The value can be changed to increase or decrease the percentage of reads that are thrown out.
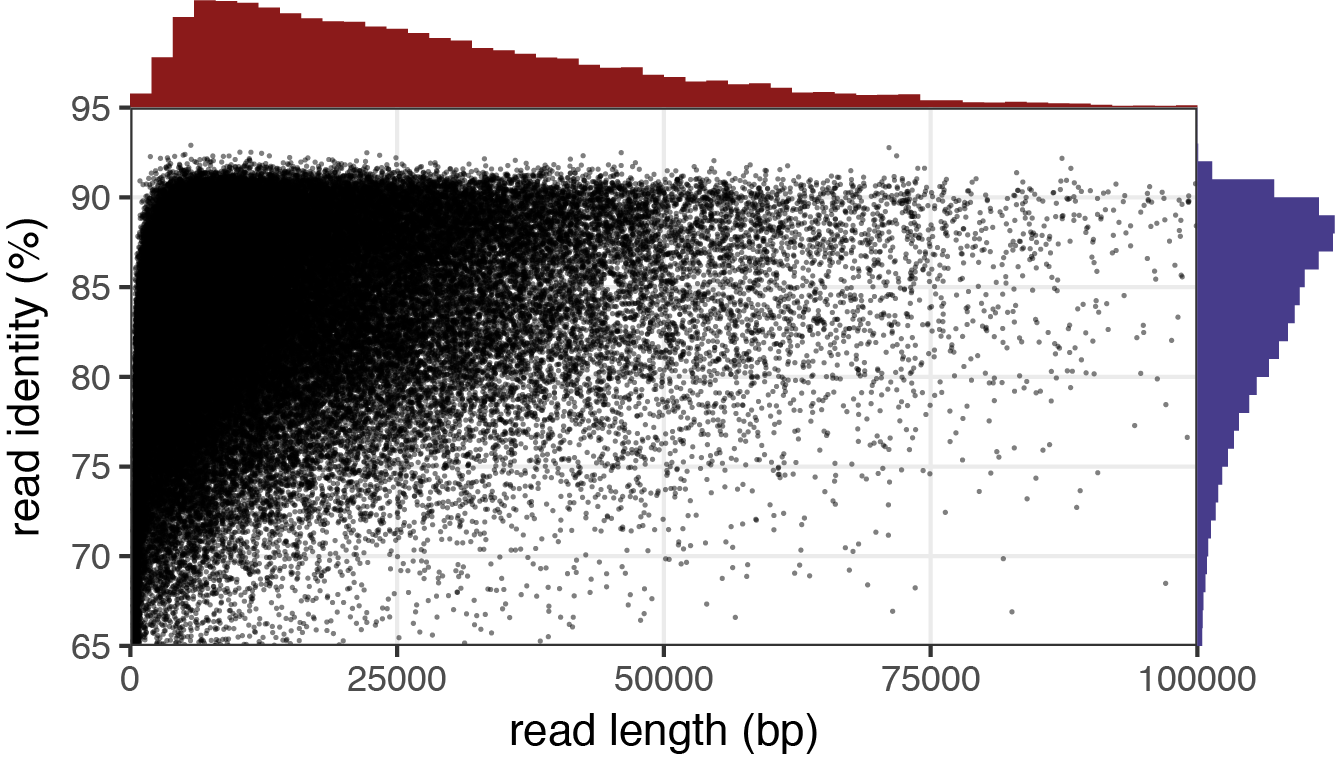
After Filtlong has run, the quality of the sequences is improved for downstream analysis. Filtlong provides the following example of 1.3 Gbp of reads before and after it was filtered using the target_bases argument to reduce the data to 0.5 Gbp of reads.
Before
After
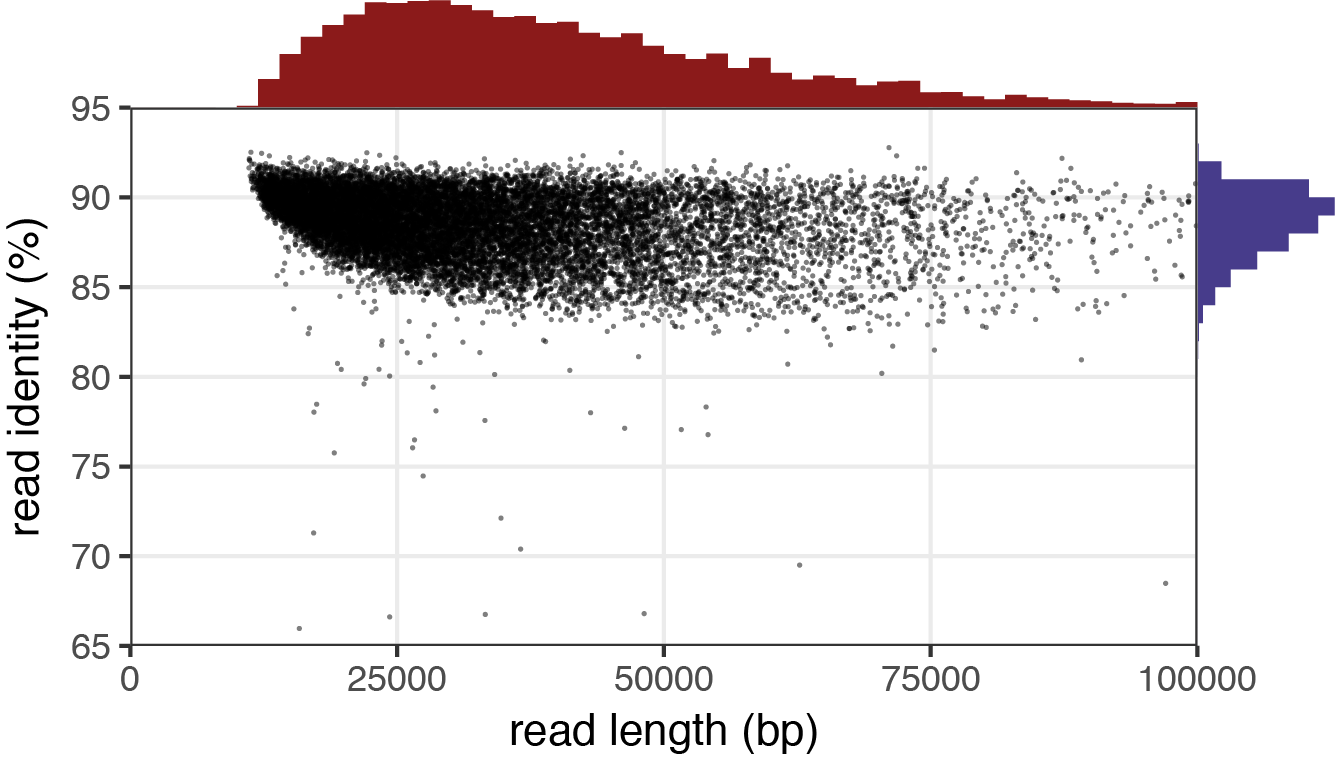
Reads with low identity percentages were removed from the dataset, leaving fewer reads with a much higher quality. Finally, we can use Nanoplot to view our results. You can read more about Filtlong and some of its other arguments on its github site: [Filtlong]https://github.com/rrwick/Filtlong
Producing reports with Nanoplot
We can produce a report that summarizes our results using the application Nanoplot. We are going to create our own before and after reports to view how Porechop and Filtlong have changed our sequencing data for downstream analysis. First, we will produce a report for our filtered reads.
Let’s take a look at the script:
sbatch nanoplot_filtered.sh
sbatch nanoplot.sh
cat /blue/general_workshop/share/bash_files/nanoplot_filtered.sh
#!/bin/bash
#SBATCH --job-name=nanoplot
#SBATCH --account=general_workshop
#SBATCH --qos=general_workshop
#SBATCH --mail-type=END,FAIL
#SBATCH --mail-user=USERNAME@ufl.edu
#SBATCH --ntasks=1
#SBATCH --mem=8gb
#SBATCH --time=12:00:00
#SBATCH --output=nanoplot_%j.out
pwd; hostname; date
module purge
module load nanoplot
NanoPlot --fastq ./Suwannee2/Suw2_filtered_3000bp_60X.fastq \
-o ./Suwannee2/Nanoplot_Suw2_filtered_out -t 1
Let’s review the options above:
--fastqis the input file type followed by its location. Other file types are supported, such asfasta,fastq_rich,bam,ubam,cram,pickleand more.-ois the output location and file name.-tis the number of threads, if additional CPUs are allocated you can increase this value.
We have included another script that produces a report from the reads prior to sending them to Porechop and Filtlong, but it is identical except the input source and output directory are different. We won’t view this script now, but if you want to view it later you can view it using the following command:
cat nanoplot.sh
The script produces an html file along with associated image files and data. Since the shell cannot render graphics, we will download all of the contents of this file to your desktop and load the html file to view the results.
First, open the tab that first appeared when you logged into UFRC On Demand and click on files in the top lefthand corner, then click on /blue/general_workshop/ from the menu:
A new screen will load. Click on Go To... at the top left, which will load a text entry box.
Now enter the following location: /blue/general_workshop/share/Suwannee/NanoplotOUT
And also check out: ‘/blue/general_workshop/share/Suwannee/Nanoplot_Suw2_filtered_out’
Click on the directory “Nanoplot_Suw2_filtered_out” and then click on the download button:
Also click and download the directory “NanoplotOUT”, which contains the unfiltered reads.
Now you can cick on the file “NanoPlot-report.html” contained in each file to view the report and the corresponding plots. Load both files. Can you see the differences between them?
Key Points
Guppy converts the unreadable electrical signals to actual nucleotide data.
Porechop removes the adapter sequences so we only view nucelotide data from our reads.
Filtlong filters reads by their length and quality.
NanoPlot produces reports that summarize our data.